














HTML
-
Porcine reproductive and respiratory syndrome (PRRS) has caused substantial economic losses to the modern swine industry since it was first reported in the United States and Canada in 1987 (Snijder and Meulenberg 1998). Reproductive failure and preterm birth in female pigs, and respiratory disease of variable severity in piglets and fattening pigs are characteristics of PRRS (Snijder and Meulenberg 1998; Lunney et al. 2016). Porcine reproductive and respiratory syndrome virus (PRRSV), the causative agent of PRRS, is an enveloped, positive-stranded RNA virus that belongs to the family Arteriviridae within the order Nidovirales (Wensvoort et al. 1992). On the basis of genetic differences, PRRSV isolates have been divided into two major genotypes, the European type (type 1) and the North American type (type 2) (Allende et al. 1999; Yuan et al. 2004). The genome of PRRSV is approximately 15.4 kb and encodes at least 10 overlapping open reading frames (ORFs) that code for eight structural proteins and 14 nonstructural proteins (Tian et al. 2007; Zhao et al. 2016).
A diverse range of Fc receptors (FcRs), widely expressed on the surface of immune cells, can bind specifically to the Fc fragment of antibodies and promote the activation of immune cells and immune complexes to trigger and regulate the immune response after formation of the FcR-Fc complex (Park et al. 1984; McCann et al. 2010). FcRs can be divided into FcγR IgG receptors (including FcγRI, FcγRIIA, FcγRIIB1, FcγRIIB2, FcγRIIIA and FcγRIIIB), FcεR IgE receptors (including FcεRI and FcεRII), FcαR IgA receptors and FcδR IgD receptors based on the different ligands they bind (Qiao et al. 2010). Activation of IgE and FcεR is the main cause of type Ⅰ allergic reactions (Suzuki et al. 1998). FcεRs can be divided into highaffinity FcεRI and low-affinity FcεRII. FcεRI has two forms, tetrameric (αβγ2) and trimeric (αγ2); the tetrameric form is expressed on mast cells and basophils while the trimeric form is expressed on other cell types (Chen et al. 1981). The α-subunit is a type Ⅰ integral membrane protein that contains two extracellular immunoglobulin domains, a transmembrane domain and a short intracellular tail, which can combine with IgE (Dhaliwal et al. 2017). Human CD23 also mediates binding of the IgE Fc domain, a feature attributed to the intrinsic flexibility of the Cε3 domain of IgE (Bournazos and Ravetch 2017). The β-chain serves to amplify receptor signal transduction (Kimberly et al. 2002). A pair of γ-chains possessing ITAM motifs can initiate IgE-triggered signaling (Wilson et al. 2002; Terada et al. 2016).
Proinflammatory cytokines (TNF-α, IL-1 and IL-6) are induced after infection by PRRSV (Nedumpun et al. 2017). PRRSV tends to attack the lungs and lymphoid organs and can proliferate in a variety of cells, such as porcine alveolar macrophages (PAMs), bone marrow-derived dendritic cells (BMDCs) and MARC-145 cells (Duan et al. 1997; Gao et al. 2016; Zhang et al. 2016). As the major host cell, alveolar macrophages expressing many receptors, such as FcγRs and FcεRs, are regarded as an efficient path to prevent PRRSV infection (Prather et al. 2017). FcεRI is an important receptor that mediates the inflammatory response (Peterfy et al. 2008). Recently, it was reported that FcγRs and a specific antibody against PRRSV could promote the virus to invade cells (Qiao et al. 2011). A systemic inflammatory response and complex clinical signs occur during PRRSV infection (Rascon-Castelo et al. 2015). Little is currently known about changes in the expression of FcεRI on lung PAMs and their effects during PRRSV infection (Rascon-Castelo et al. 2015). Understanding the role of porcine FcεRI and its expression changes during PRRSV infection will help in understanding the complex inflammation response and pathogenic mechanisms of PRRSV.
-
First-strand cDNA was synthesized from purified RNAs of PAMs and 3D4/21 cells using a First-Strand Synthesis System (Transgen, Beijing) according to the manufacturer's instructions. The FcεRI gene was subsequently amplified using primers FcεRI-F and FcεRI-R, based on known sequences of FcεRI genes, as shown in Supplementary Table S1, and the amplified fragments were cloned into pGEM®-T Easy Vector (Transgen). The vector containing the FcεRI gene was sent to Genewiz (Beijing, China) for sequencing. Enzymes used for cloning procedures were purchased from TaKaRa (Dalian, China). A DNA thermal cycler (Biometra Tgradient, Germany) was used to perform the reaction. The FcεRI gene sequence was deposited at NCBI (GenBank Number: XM_001929139).

Table Table S1. The primers used for PCR amplification
-
The pcDNA3.1-FcεRI eukaryotic expression vector was constructed. Primers pcDNA3.1-FcεRI-F and pcDNA3.1- FcεRI-R (Supplementary Table S1), harboring common sequence with the vector (underlined), were used to amplify the FcεRI gene from the FcεRI clone plasmid. Through the common sequence, the PCR product was ligated with pcDNA3.1 vector by using a one-step cloning kit (Vazyme, China). The prokaryotic expression plasmid pGEX-6p-1 was constructed using primers pGEX-6pFcεRI-F and pGEX-6p-FcεRI-R, to express the recombinant protein GST-FcεRI according to the aforementioned protocol.
-
PAMs and PBMCs were collected from PRRSV-negative piglets from Tianjin Ninghe original pig farm according to a protocol described previously (Arce et al. 2010; Sinha et al. 2012; Uddin et al. 2012). The intestinal porcine epithelial cell line IPEC-J2 and the porcine kidney cell line PK-15 were purchased from the Chinese Veterinary Drugs Inspection Institute (Beijing, China). All applicable international, national and/or institutional guidelines for the care and use of animals were followed. Pigs were maintained in individually ventilated cages at the Tianjin Laboratory Animals Center.
PRRSV-permissive derivative 3D4/21 cells (PAMpCD163, obtained by transfecting the viral receptor CD163 into CRL2843 AKA 3D4/21 cells), were kindly contributed by Han Jun, professor of China Agricultural University (Lee et al. 2010). 3D4/21 cells were cultured in RPMI 1640 medium (Gibco, USA) supplemented with 10% (V/V) fetal bovine serum (FBS) plus 100 μg/mL penicillin and streptomycin, were used to inoculate and propagate PRRSV. Human embryonic kidney (HEK) 293T cells, IPEC-J2 and PK-15 were maintained in Dulbecco's modified eagle's medium (DMEM, Gibco) supplemented with 10% (V/V) FBS plus 100 μg/mL penicillin and streptomycin. All cells were cultured in a humidified incubator with 5% CO2 at 37 ℃.
PRRSV (JXwn06) were contributed from China Agricultural University. PRRSV was propagated in 3D4/21 cells, and the PRRSV titer was determined to be 106.54 TCID50/0.1 mL using the Reed-Muench method (Reed and Muench 1938). Viruses were frozen at - 80 ℃ before use.
-
PRRSV-specific IgG (IgG+) and specific-pathogen-free pig serum (PRRSV-negative IgG, marked as IgG-) were identified using an IDEXX HerdChek PRRS X3 Ab enzyme-linked immunosorbent assay (ELISA) (Westbrook, MA), and purified antibodies were further precipitated by using saturated ammonium sulfate to yield a protein content of 8.160 and 7.92 mg/mL, respectively. Anti-b-actin mouse monoclonal antibody and goat anti-mouse IgG (H+L) horseradish peroxidase conjugate were purchased from TIANGEN (Beijing, China).
The polyclonal antibody against FcεRI was derived from serum of rabbit immunized with extracellular recombinant GST-FcεRI proteins coupled with mineral oil adjuvant as previously described (Zhang et al. 2016).
The ELISA plate (Bio-Rad, USA) was coated with the recombinant FcεRI protein at 4 ℃ overnight, and then blocked with 5% horse serum (Gibco, USA). After washing three times with PBS, the diluted FcεRI antiserum (with the dilution ratios of 1:50, 1:100, 1:200, 1:400, 1:800, 1:1600, 1:3200, 1:6400, 1:12, 800 and 1:25, 600) was added to the corresponding well and incubated at 37 ℃ for 1 h. Subsequently, the plates were washed with PBS and incubated with enzyme-labeled secondary antibody Goat-Anti-Rabbit-HRP (CWBIO, Beijing) diluted 1:5000. Then add freshly prepared OPD substrate coloring solution and stop solution to the corresponding well. Absorbance was measured at 490 nm using a microplate reader (Thermo Fisher, USA) within 15 min after termination.
The immunofluorescence assay was used to detect FcεRI expression. HEK 293T Cells were transiently transfected with 1 μg plasmid of pcDNA3.1-FcεRI or pcDNA3.1 for 24 h, The transfected cells were fixed and the rabbit anti-FcεRI polyclonal antibody or rabbit FcεRInegative antibody (from PBS-immunized rabbit) were added to react at 37 ℃ for 30 min, and then the goat antirabbit FITC conjugate (Abcam, UK) was added at 1000-fold dilution for 30 min, and the nucleus was stained with 40, 6-Diamidino-2-Phenylindole (DAPI). The localization of FcεRI were observed under an inverted confocal microscope (PerkingElmer, UltraView Vox, USA). Images were taken at a ×100 magnification.
-
The purified IgG+ or IgG- with the concentration of 6.25 μg/mL was mixed with 1 MOI of PRRSV, and incubated on ice for 1 h to promote the formation of virusantibody immune complexes.
The 3D4/21 cells were seeded into 6-well plates and inoculated with 1 MOI of PRRSV, 1 MOI of PRRSV and 200 μL negative IgG (PRRSV + IgG-), 1 MOI of PRRSV and 200 μL positive IgG (PRRSV + IgG+) or phosphatebuffered saline (PBS) respectively at 4 ℃ for 1 h. After 24 h, cells were harvested and the FcRs mRNA expression level were analyzed by quantitative reverse transcription PCR (RT-qPCR).
The 3D4/21 cells or HEK 293T were transfected with pcDNA3.1-FcεRI or pcDNA3.1 (1 μg) with 5 μL PEI (Head, Beijing) respectively following the manufacturer's instructions. After 24 h, the layer cells were respectively inoculated with PRRSV, PRRSV + IgG-, PRRSV + IgG+ or PBS at the previous dose. After 24 h, the cells were harvested and subjected to analyze the PRRSV RNA load level and the expression level of inflammatory- related genes by RT-qPCR.
-
Total cellular RNA was extracted from cells using TRIzol, and complementary DNAs (cDNAs) were synthesized using a First-Strand Synthesis System (Transgen, Beijing) according to the manufacturer's instructions.
The amplification reaction contained 2 × Multiplex PCR Mix (10 μL), 10 mmol/L forward and reverse primers, 2 μL of the cDNA template, and sterile double-distilled water to a final volume of 20 μL. All RT-qPCRs were performed under the following conditions: 95 ℃ for 5 min followed by 40 cycles of 95 ℃ for 30 s and 56 ℃ for 30 s. SYBR Green Master Mix (Vazyme) was used according to the manufacturer's instructions, and the real-time PCR was performed on a 7500 Real-time PCR system (Applied Biosystems, Foster city, CA, USA). All data presented were quantified relative to the mRNA level of the endogenous gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and analyzed using GraphPad Prism 6.0 software (La Jolla, CA 92037 USA). The primer pairs used for RT-qPCR are shown in Supplementary Table S2.

Table Table S2. The primers used for RT-qPCR detection
-
HEK 293T cells transfected with pcDNA3.1-FcεRI or pcDNA3.1 (1 μg) with 5 μL PEI (Head, Beijing) respectively following the manufacturer's instructions. Twentyfour hours after transfection, cells were washed twice with PBS and harvested in RIPA buffer. Cell lysates were heated in buffer for 10 min and separated by SDS-PAGE. The separated proteins were transferred onto a nitrocellulose fitter membrane (NC) (PALL, USA), and then membranes were blocked with PBST (1 × PBS in 0.05% Tween 20) and 5% skim milk for 1 h at room temperature. The membranes were then stained with primary antibodies at 4 ℃ overnight. The membranes were washed thrice with PBST for 3–5 min in each interval in order to remove excessive antibodies, and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 2 h. To detect the bioactivity of FcεRI polyclonal antibody, the primary antibodies are GST monoclonal and FcεRI polyclonal antibody. To detect the FcεRI expression in transfected HEK 293T cells, the primary antibodies are the Myc antibody (TransGen, Beijing) (1:5000) and β-actin antibody (TransGen, Beijing) (1:5000).
Signals were visualized using Pierce ECL Western Bloting Substrate (Thermo Scientific), and images were obtained using a chemiluminescence apparatus (Gel Doc XR + imaging system; Bio-Rad, USA).
-
One million 3D4/21 monolayer cells were inoculated respectively with 1 MOI of PRRSV, 1 MOI of PRRSV and 200 μL negative IgG (PRRSV + IgG-), 1 MOI of PRRSV and 200 μL positive IgG (PRRSV + IgG+) or PBS. The cells were collected and washed with PBS after 24 h of inoculation, resuspended in 1 mL TRIzol LS (Invitrogen, USA) and transferred to cryopreservation tubes. Transcript mRNA was obtained by using oligo (dT)18 enrichment mRNA that was fragmented, purified, and subjected to mass detection. A terminal sequence was ligated, and the cDNA was amplified by reverse transcription before using a machine for high-throughput sequencing. All samples were sent to Guangzhou GENE DENOVO (Guangzhou, China) for transcriptome sequencing.
-
The above experiments were carried out with three independent replicates, and the data were analyzed using the 2-ΔΔCT method. Data were expressed as mean ± SD. Pairwise multiple comparisons were conducted to determine differences between groups by two-way analysis of variance (ANOVA) followed by Bonferroni post-tests using GraphPad Prism 6.0 (GraphPad Software Inc.). P-values < 0.05 were considered statistically significant.
Cloning of the FcεRI Gene and Sequencing
Plasmid Construction
Cells and Viruses
Generation and Characterization of Antibodies
Infection and Transfection
Quantitative Reverse Transcription PCR (RT-qPCR)
Western Blotting
Sequencing the Transcriptome of 3D4/21
Statistical Analysis
-
To determine the expression changes of porcine gamma and epsilon Fc receptors caused by PRRSV inoculation or PRRSV-antibody complex infection, a 3D4/21 cell infection model was used. In the case of PRRSV infection alone, only the expression of FcγRI was enhanced remarkably, and the change in expression of the other receptors was not significant. As shown in Fig. 1, the transcriptional level of FcγRI was slightly increased, while that of FcγRIIb (inhibitory receptor) was significantly enhanced, in the case of infection with the PRRSV-antibody complex (PRRSV + IgG+). At the same time, the transcription level of FcεRI was increased remarkably, while the effect on FcεRII was little. We speculated that the PRRSV-antibody immune complex may influence the phenotype of PAMs and induce FcεRI overexpression through some pathway. So we mainly considered FcεRI in the following experiments.
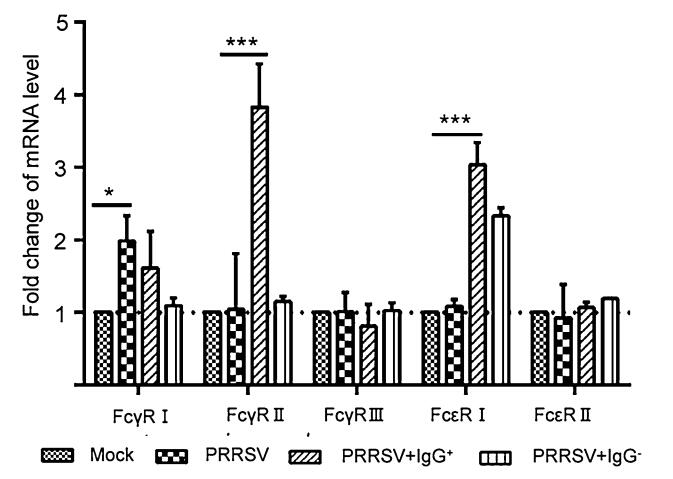
Figure 1. Effect of PRRSV on mRNA expression level of different FcRs in 3D4/21 cells. The purified IgG+ or IgG- with the concentration of 6.25 μg/mL was mixed with 1 MOI of PRRSV, and incubated on ice for 1 h to promote the formation of virus-antibody immune complexes. The 3D4/21 cells were seeded into 6-well plates and inoculated with PRRSV, PRRSV + IgG+, PRRSV + IgG- and PBS respectively. Cells were harvested and subjected to RT-qPCR analysis 24 h after PRRSV infection. Bars indicate the 2-ΔΔCT of FcR mRNA copies in infected cells or mock-infected cells. Error bars indicate the SD from three independent experiments. ***P < 0.001, **P < 0.01, *P < 0.05.
-
Using oligonucleotide primers based on sequences of porcine FcεRI in NCBI GenBank and template cDNAs from PAM cells, we obtained fragments of approximately 800 base pairs by PCR (Supplementary Fig. S1A). For further experiments, these fragments were cloned into the vector pGEM®-T Easy (Transgen), which could be replicated in Escherichia coli DH5α. The cloned FcεRI gene was sequenced, and no frame-shifts or mutations were detected in the coding sequence. The nucleotide sequence of the cloned FcεRI (GenBank Number: KU527059) was very similar to those from GenBank, with 99.86% identity by sequence alignment, so we confirmed that FcεRI was successfully cloned from PAM cells.

Figure Supplementary Figure S1. Cloning and analysis of the porcine FcεRI gene. (A) Cloning of the FcεRI gene from PAMs. Lane M, DNA molecular weight marker; lane 1, FcεRI obtained from the PAMs. (B) Cloning of the FcεRI gene from 3D4/21, PBMC, PK-15 and IPEC-J2 cells. Lanes: M, DNA molecular weight marker; 1, 3D4/21 cells; 2, PBMC cells; 3, PK-15 cells; 4, IPEC-J2 cells; 5, negative control.
Subsequently, RT-PCR was performed to detect FcεRI expression from total RNA extracted from 3D4/21, PBMC, PK-15 and IPEC-J2 cells (Supplementary Fig. S1B). In contrast to the 3D4/21 cell line, FcεRI was not detected in PBMC, PK-15 or IPEC-J2 cells. Besides, the fragment size of the sequence amplified from the 3D4/21 cells was consistent with the expected size. It was demonstrated that 3D4/21 could express FcεRI in high abundance but the other cell lines could not.
The biological activity of rabbit anti-porcine FcεRI polyclonal antibody was evaluated by ELISA, Western blot and cell receptor reactivity detection. The ELISA titer of FcεRI polyclonal antibody was 1:12, 800 (Fig. 2A). The FcεRI polyclonal antibody had good reactivity with recombinant FcεRI protein (Fig. 2B) as detected by Western blot. HEK 293T cells were transfected with pcDNA3.1-FcεRI plasmid, and a fluorescent antibody was used to analyze the specificity of the FcεRI polyclonal antibody (Fig. 2C). The rabbit anti-porcine FcεRI polyclonal antibody specifically recognized FcεRI expressed on HEK 293T cells (Fig. 2C-a).

Figure 2. Generation and characterization of recombinant FcεRI polyclonal antibody. A Antibody titers were measured by indirect ELISA. B The effectiveness of polyclonal antibodies was verified by Western blot experiments. Lanes: M, protein molecular weight marker; 1, reactivity with GST monoclonal antibody; 2, reactivity with FcεRI polyclonal antibody. C Specificity and bioactivity detection of FcεRI polyclonal antibody. HEK 293T cells were transfected with plasmids pcDNA3.1-FcεRI (C-a, C-c) or control vector pcDNA3.1 (C-b, C-d). At 24 h, cells were fixed and then subjected to immunofluorescence assay to detect FcεRI expression using rabbit anti-FcεRI polyclonal antibody (C-a, C-b) and rabbit negative antibody (C-c, C-d). The nucleus is stained with DAPI (blue) in the merged images.
-
The PRRSV titers in FcεRI-overexpressing cells and cells treated with an anti-FcεRI antibody were detected to evaluate the role of FcεRI in 3D4/21 cells. As shown in Fig. 3A, the mRNA level of PRRSV was significantly increased after overexpression of FcεRI, which suggested that FcεRI may promote engulfment of the PRRSV-antibody complex into cells to enhance PRRSV infection. On the contrary, in the cells in which FcεRI was blocked by the antibody, the proliferation of PRRSV was inhibited (Fig. 3B).

Figure 3. FcεRI is involved in PRRSV-antibody immune complex attachment and internalization. A 3D4/21 cells were transfected with pcDNA3.1-FcεRI or pcDNA3.1 (1 μg) for 24 h, respectively, and then the transfected cells were treated with PRRSV, PRRSV + IgG-, PRRSV + IgG+ (MOI = 1) or PBS (Mock). Twenty-four hours after PRRSV infection, cells were harvested. PRRSV RNA load levels were analyzed by RT-qPCR. B 3D4/21 cells were treated with rabbit anti-FcεRI antibody against FcεRI (to block FcεRI) in an incubator at 37 ℃ for 1 h, and then treated with PRRSV, PRRSV + IgG-, PRRSV + IgG+ (MOI = 1) or PBS (Mock). Twenty-four hours after PRRSV infection, cells were harvested. PRRSV RNA load levels were analyzed by RT-qPCR. C Western blotting analysis of FcεRI protein in HEK293-T cells. D HEK 293T cells were transfected with pcDNA3.1-FcεRI or pcDNA3.1 (1 μg) for 24 h, respectively, and then the transfected cells were treated with PRRSV, PRRSV + IgG-, PRRSV + IgG+ (MOI = 1) or PBS (Mock). Twenty-four hours after PRRSV infection, cells were harvested. PRRSV RNA load levels were analyzed by RT-qPCR. Bars indicate the 2-ΔΔCT of FcεRI mRNA copies in infected cells or mock-infected cells; error bars indicate SD from two independent experiments. *P < 0.05 according to t test.
More than one type of FcR is expressed on the surface of PAMs (Liu et al. 2010). Both porcine FcγRI and FcγRIIb receptors play crucial roles in the ADE of PRRSV (Gu et al. 2015). Whether or not FcεRI mediates the attachment of the PRRSV-antibody immune complex and further facilitates the multiplication of PRRSV is still unclear. We investigated the effect of FcεRI alone by transfecting a non-FcεRI-bearing cell line (HEK 293T). As shown in Fig. 3C, the pcDNA3.1-FcεRI plasmid could be expressed highly in HEK 293T cells. Then, HEK 293T were transfected with pcDNA3.1-FcεRI or pcDNA3.1, and treated with PRRSV, PRRSV + IgG-, PRRSV + IgG+ or PBS. As shown in Fig. 3D, no PRRSV RNA was detected in the nonsensitive HEK 293T cells after PRRSV infection alone. But the FcεRI-transfected HEK 293T cells became susceptible to PRRSV multiplication after pretreatment with PRRSV-antibody complex. This result suggested that FcεRI could mediate attachment of the PRRSV-antibody immune complex to its target cells, and that might contribute to virus internalization and subsequent replication.
-
The above-mentioned data hints that FcεRI may participate in signal transduction and antigen presentation processes after infection by PRRSV alone or as PRRSV-antibody complexes. In both cases, the transcription of genes encoding virus recognition receptor (TLR7), antigen presenting molecules (MHCII) and inflammatory cytokines (MCP-1, IFN-β, TNF-α), biomarker molecules on PAM cells (CCR2), and signal transduction transcription factors (NFκB, p38) was examined to understand the regulatory role of FcεRI in virus infection of cells. 3D4/21 cells were transfected with pcDNA3.1-FcεRI or pcDNA3.1 for 24 h or treated with antibody against FcεRI in an incubator at 37 ℃ for 1 h, and then the transfected cells were treated with PRRSV, PRRSV + IgG-, PRRSV + IgG+ or PBS (MOI = 1). Twenty-four hours after PRRSV inoculation, cells were harvested and subjected to analysis of cytokine mRNA levels by RT-qPCR. As shown in Fig. 4A, the expression of MHCII was significantly enhanced under FcεRI overexpression, but no significant change was observed in the other experimental groups. The results illustrated that antigen presentation through the MHCII process was prompted by high FcεRI expression during PRRSV infection.

Figure 4. Fold change of cytokine mRNA levels in PRRSV-infected 3D4/21 cells. 3D4/21 cells were transfected with pcDNA3.1-FcεRI or pcDNA3.1 (1 μg) for 24 h, and then the transfected cells were treated with PRRSV, PRRSV + IgG-, PRRSV + IgG+ (MOI = 1) or PBS (Mock). For the antibody block, 3D4/21 cells were treated with antibody against FcεRI in an incubator at 37 ℃ for 1 h, and then treated with PRRSV, PRRSV + IgG-, PRRSV + IgG+ or PBS. Twenty-four hours after PRRSV inoculation, cells were harvested and subjected to analysis of cytokine mRNA levels by RT-qPCR. A MHCII mRNA levels, B TLR7 mRNA levels, C TNF-α mRNA levels, D IFN-β mRNA levels. Bars indicate the 2-ΔΔCT of cytokine mRNA copies in infected cells or mock-infected cells; error bars indicate SD from three independent experiments. ***P < 0.001, **P < 0.01, *P < 0.05.
To investigate the cross-interference relationship between FcεRI and other receptor pathways, we detected the transcriptional level of the Toll-like receptor 7 (TLR7) and some cytokines. TLR7, belonging to the patternrecognition receptor (PRRs), can induce the production of antiviral cytokines (Du et al. 2016). The transcription level of TLR7 after PRRSV infection was enhanced significantly in the FcεRI-overexpression group, and was 3–4 times higher than that in the antibody-blocking group (Fig. 4B). The increased virions in infected cells generated by the FcR routine, and cross-talk or cross-interference between the TLR and FcR pathways might contribute to the activation of TLR7; the mechanism needs be further studied. The transcript levels of TNF-α and IFN-β in the PRRSVinfected cells were up-regulated significantly after transfection with FcεRI, and no other effects were observed in the other treatments such as the cells with FcεRI blocked (Fig. 4C, 4D). The up-regulated FcεRI increased the levels of proinflammatory cytokines TNF-α and IFN-β, which indicated that FcεRI plays a leading role in the inflammatory response and viral immune response (Dong et al. 2015; Auray et al. 2016).
In order to further study the regulation of FcεRI in PRRSV + IgG- and PRRSV + IgG+ infection, we analyzed the transcriptome sequencing data and found enrichment of genes associated with FcεRI-mediated signaling pathways, inflammatory factors and inflammation. Multiple signaling pathways were activated during PRRSV + IgG- infection (Fig. 5A). Transcript levels of genes encoding Ras, p38 (Yao et al. 2017) and other inflammatory factors and TNF-α were down-regulated, especially in the PRRSV + IgG+ experimental group (Fig. 5A); those of genes encoding AKT (Yao et al. 2017), ERK (Guo et al. 2017; Yao et al. 2017), MKK3/6, Vav and other signal molecules showed a significant increase, thus promoting smooth muscle contraction and recruitment of effector cells, causing a subsequent anti-inflammatory response (Fig. 5A). Some inflammatory regulatory molecules showed variable changes during PRRSV infection. The expression levels of genes encoding PLA2G6, LOX, TRPV1, ASIC1, GSG2, TRPM8 (Kozyreva et al. 2016) and TRPM4 were significantly up-regulated during PRRSV + IgG- infection, while the expression levels of those cytokines showed a more significant down-regulation under PRRSV + IgG+ infection, indicating that the inflammatory response was indeed caused during PRRSV/IgG- infection (Fig. 5A). As shown in Fig. 5A, genes encoding the anti-inflammatory cytokines IL-1R, IL-15, CCL-20, TGF-β (de Witte et al. 2017), FAS (Khan et al. 2017) and NF-κB exhibited down-regulation under the PRRSV-ADE effect (PRRSV + IgG+ infection). Unbalancing of inflammatory regulatory molecules, inflammatory cytokines and anti-inflammatory cytokines caused NFκB pathway activation and a more intense inflammatory cytokine (MCP-1) production. A quantitative fluorescence method was used to detect thesignal pathway-related molecules p38 and NF-κB, and the results were consistent with those of the transcriptome analysis (Fig. 5B).

Figure 5. Expression of inflammatory-related genes in 3D4/21 cells. A 3D4/21 cells were infected with PRRSV + IgG-, PRRSV + IgG+ or PBS. After 24 h, Cells were harvested and subjected to RT-qPCR anlaysis. Heat map of inflammatoryrelated receptor genes was constructed using the Morpheus heat map building tool (https://software.broadinstitute.org/morpheus/). B 3D4/21 cells were infected with PRRSV + IgG-, PRRSV + IgG+ or PBS (Mock) at an MOI of 1 and then harvested 24 h post-infection and subjected to qRT-PCR analysis. Bars indicate the 2-ΔΔCT of mRNA copies of chemokines or transcription factors in infected cells or mock cells; error bars indicate SD from three independent experiments. ***P < 0.001, **P < 0.01, *P < 0.05.
Effect of PRRSV Infection on FcR Transcription in 3D4/21 Cells
Generation and Characterization of FcεRI Polyclonal Antibodies
FcεRI Enhances Viral Replication During PRRSV Infection
Regulation of Inflammation by Porcine FcεRI in PRRSV Infection
-
Although vaccines against PRRSV have been developed and used widely, PRRS has brought an enormous economic loss to the porcine industry around the world (Huang et al. 2009; Wang et al. 2016). Antibodies induced by the vaccines can prevent PRRSV infection based on antibody neutralization, while vaccine-induced antibodies may also induce the antibody-dependent enhancement (ADE) of PRRSV (Gu et al. 2015). It was reported in 1993 that PRRSV infection can be enhanced by the PRRSV-antibody immune complex (Christianson et al. 1993). Evidence was then provided that the ADE of PRRSV infection in vivo was induced by PRRSV-specific IgG (Yoon et al. 1996; Yoon et al. 1997). In accordance with these findings, we found that PRRSV-specific antibody prevented virus infection in PAMs at a high level, but enhanced virus multiplication in PAMs at a proper antibody titer level.
The mechanism of ADE caused by PRRSV infection involves several cell-surface molecules, including the FcRs (Halstead and O'Rourke 1977), complement receptors, CD4/15/33 (Novakovic et al. 2016; Yuste et al. 2017) and b2-microglobulin (Tirado and Yoon 2003; Bao et al. 2013). The antibody-FcγR interaction is known to play an important role in the ADE phenomenon (Zhang et al. 2016). The suggestion that FcγR can mediate ADE in dengue virus infection was first made in 1977 (Halstead and O'Rourke 1977).
We found that the transcriptional level of FcγRIIb and FcεRI was increased remarkably in our research (Fig. 1). According to a previous study, the porcine FcγRIIb receptor plays crucial roles in the ADE of PRRSV. However, whether FcεRI mediates the attachment of the PRRSV-antibody immune complex and further facilitates the multiplication of PRRSV is still unclear. Obvious anaphylaxis-like clinical signs and inflammatory responses were observed in PRRSV-infected pigs (Qiao et al. 2011), and FcεRI can induce the production of generous inflammatory mediators in the type Ⅰ allergic reaction (Nagai et al. 1986). Although direct evidence of FcεR-mediated ADE has not been reported, we initially assumed that FcεRI may mediate ADE of PRRSV infection.
FcεRI was first cloned and characterized from mast cells and basophils (Daëron et al. 1985). Two types of FcεRI have been reported: one is a tetrameric αβγ2 form on mast cells and basophils and the other one is a trimeric αγ2 form on many other cell types (Chen et al. 1981). The α-subunit is the main domain that combines with IgE (Platzer et al. 2011; Takayama et al. 2013). In our study, the porcine FcεRI α-chain was detected and cloned from primary PAMs and the monocyte-macrophage line 3D4/21. In addition, as the target cells of PRRSV infection, massive PAMs highly and dynamically express the FcεRI receptor on their cell surface. The role of FcεRI in inflammatory responses caused by PRRSV infection should be discussed and further studied.
Owing to the fact that multiple FcRs are expressed on the surface of PAMs, pcDNA3.1-FcεRI was transfected into HEK 293T cells, a PRRSV non-sensitive cell line (Qiao et al. 2009), to get FcεRI-positive cells, and the PRRSV titer was monitored from the FcεRI-positive HEK 293T cells after inoculation with the PRRSV-antibody complex to understand the function of FcεRI. The result suggested that FcεRI may enhance the intake of PRRSV through the pathway mediated by FcεRI and promote the replication of PRRSV. At the same time, 3D4/21 cells, a permissive cell for PRRSV, were used as receptor cells to achieve high FcεRI expression. The transcriptional level of PRRSV was higher in the group transfected with the pcDNA3.1-FcεRI plasmid than in the group in which FcεRI was blocked by an antibody and in the blank-vector treatment group. The expression of several FcγR genes can be regulated by cytokines and chemokines presenting during inflammation, tissue injury and infection throughout leukocyte development and differentiation. Alterations in the FcγR expression pattern readily influence the outcome of FcγR-mediated signaling and have profound consequences on the immunomodulatory functions initiated upon FcγR crosslinking by IgG immune complexes (Bournazos and Ravetch 2017). Both activating and inhibitory FcγRs can mediate phagocytosis. Initial steps of phagocytosis following receptor oligomerization and phosphorylation include actin remodeling and cytoskeletal reorganization, ultimately leading to the endocytosis of the FcγR-IgG complex (Bournazos and Ravetch 2017). Our results hint that FcεRI may be involved in endocytosis of the FcγR-IgG complex, PRRSV multiplication and regulation of the antiviral response during PRRSV infection. Interestingly, we observed that transcriptional expression of TLR7 was increased when 3D4/21 cells were treated with PRRSV + IgG+. The increased virions entering infected cells by the FcR routine, or cross-talk and crossinterference between the TLR and FcR pathways, might contribute to the activation of TLR7; however, the mechanism needs be further studied.
Indeed, under physiological conditions, FcεRI could be detected in PAM cells by fluorescence quantitative PCR, and the expression level was no less than in PAM cells infected by PRRSV. However, the expression was greatly promoted in PAM cells treated with PRRSV-antibody complex before inoculation with PRRSV.
As noted above, the mRNA levels of signaling pathway molecules p38 and NF-κB showed down-regulation, especially in the PRRSV + IgG+ infection groups. At the same time, some of the signal molecules downstream, such as AKT, ERK (a direct reflection of the occurrence of inflammation), PLA2G6 (Chew and Ong 2016), LOX (Neau et al. 2014), TRPV1 and other molecules that regulate inflammation increased under the PRRSV-ADE effect. What's more, the down-regulation of genes encoding the anti-inflammatory molecules IL1R, CCL-20, TGFb and FAS also indicated that PRRSV infection can cause a serious inflammatory response. In conclusion, we have shown that FcεRI influences antiviral responses and is involved in the ADE of PRRSV infection. These findings expand our understanding about the mechanism of FcεRI in mediating the ADE phenomenon, and may provide a new method to prevent PRRSV infection.
-
This work was supported by the National Natural Science Foundation of China (31272540) and the underprop project of Tianjin Science and Technology Committee in China (16YFZCNC00640).
-
JH conceived and designed the experiments. PS, JW, DL, MS, Lilin Zhang, YL, Lei Zhang performed the experiments. JW and JR analyzed the data. JW and JH wrote the paper. JH, Lei Zhang checked and finalized the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Author Contributions
-
The authors declare that they have no conflict of interest.
-
Animal experiments were performed in compliance with the regulations set by Tianjin University Institutional Animal Care and Use Committee and were approved by the veterinarian authorities of Tianjin Animal Inspection Institute. This study was carried out in strict accordance with the guidelines in the Guide for the Care and Use of Laboratory Animals in China.

 DownLoad:
DownLoad: