













HTML
-
In eukaryotic cells, the ubiquitin-proteasome pathway (UPP) plays important roles in many cellular events by targeting certain regulatory proteins for 26S proteasome degradation. This process is carried out in two consecutive steps: (1) tagging the substrate with a specific type of ubiquitin chain and (2) subsequent degradation of the tagged substrate by the 26S proteasome complex (Saeki 2017). As a highly conserved 76 amino acid polypeptide, ubiquitin is conjugated to substrates via an enzymatic cascade by activating enzyme (E1), ubiquitin-conjugating enzyme (E2) and ubiquitin ligase (E3) (Ciechanover 1994). E3s bind E2 and determine specificity of the substrate to be ubiquitinated. Some classes of E3s, such as HECT family E3s, carry the activated ubiquitin and catalyze substrate ubiquitination by themselves. Other E3s, such as RING family E3s, merely bring the substrate in close proximity to E2 while E2 directly catalyzes the transfer of ubiquitin to the substrate. Additional ubiquitin molecules can be anchored to Met1 or one of the seven lysine residues on ubiquitin to form a polyubiquitin chain. When linked through Lys48, the polyubiquitinated substrates are recognized and degraded by the 26S proteasome complex in the ubiquitin-proteasome pathway (UPP) (Komander and Rape 2012).
Owing to its critical role in regulating protein degradation, the UPP plays important role in host immunity against pathogens including viruses. The cellular deubiquitinating (DUB) USP15 was shown to induce degradation of HIV viral protein Nef and Gag, thus hampering HIV-1 replication (Pyeon et al. 2016). Host E3 TRIM41 was revealed tomediate N protein ubiquitination and degradation of vesicular stomatitis virus, leading to viral restriction (Patil et al. 2020). On the other hand, UPP is occasionally hijacked by various viruses to defeat the host immune system. HIV encodes a series of viral protein adaptors to utilize the host UPP for its own privilege. The HIV-1 Vif protein hijacks the CBF-b– CUL5–ELOB–ELOC E3 complex to target the host immune factor APOBEC3G for degradation (Guo et al. 2014). The HIV-1 Vpu protein is also reported to sequester BST-2 through interaction with the β-TrCP E3 subunit (Bego et al. 2015). The HIV-2 Vpx protein recruits the CUL4A-DDB1- DCAF1 E3 ligase to induce the ubiquitination and degradation of the immune factor SAMHD1 (Miyakawa et al. 2019). Taken together, these studies reveal the importance of UPP during viral infection and host immune response.
Since viruses are competent in hijacking the intracellular ubiquitin-proteasome system to escape from host immune responses, it would be also applicable for us to engineer cellular E3s to instruct the host UPP to target more vital viral proteins. The ideas to engineer E3s have been successfully accomplished. Proteolysis Targeted Chimeras (PROTACs) have been designed to tether a substrate binding ligand with an E3, triggering the destruction of certain intracellular proteins (Gadd et al. 2017). Ubiquibodies, synthetic E3s whose substrate recognition domains were replaced with intracellular antibody fragments, offer a reliable means to remove specific cellular proteins through UPP (Portnoff et al. 2014). The auxin-inducible degron (AID) system is designed to attach a degron derived from auxin transcription repressors to the substrate protein, which can consequently be recognized by auxin-binding E3 TIR1 upon stimulation with auxin, leading to substrate degradation (Nishimura et al. 2009). The Trim-Away method first utilizes microinjected cytosolic antibodies to bind cellular substrates, then E3 TRIM21 which recognizes Fc domain of antibody is recruited and targets the antibody-bound substrates for degradation (Clift et al. 2017). In this study, we aimed to develop chimeric E3s which target viral proteins for degradation, thus achieving improved cell protection from viral infection. Considering its virulence and lack of vaccine, human immunodeficiency virus 1 (HIV-1) has been chose to be our target for candidate viral proteins.
Although combination antiretroviral therapy (cART) has turned HIV/AIDS from an incurable disease into a treatable chronic disease, the rapid mutation rate of the HIV genome constantly challenges the clinical therapeutic effects of ART (Hare et al. 2010a, b; Feng et al. 2013). HIV viral proteins are mainly composed of structural proteins, viral-specific enzymes and accessory proteins. Their functions are highly dependent on well-studied virushost protein interactions, which are conserved in all fully functional viruses (Simon et al. 2015). Furthermore, a comprehensive database of HIV-1 human protein interactions (HIV-1 Human Interaction Database, https://www.ncbi.nlm.nih.gov/genome/viruses/retroviruses/hiv-1/interactions/) has been established, making it possible to identify regions involved in virus-host protein interactions that can be used for chimeric E3 designs.
Based on database query, a series of prototypical chimeric E3s have been designed, constructed and functional analyzed. After further optimization, chimeric E3 146LIS is generated to be capable of targeting HIV-1 NL4-3 integrase for Lys48-specific polyubiquitination and degradation by 26S proteasome. Jurkat cells with 146LIS CRISPR knock-in showed a significant reduction in HIV-1 NL4-3 viral DNA integration when challenged with infectious viruses. Simultaneously, HIV-1 NL4-3 viral replication was also suppressed. No obvious cell cytotoxicity was observed in lymphocytes with continuously expression of 146LIS.
-
Chimeric E3s and HIV-1 integrase sequences were synthesized by Tsingke Company, Wuhan, China. Sequence information was obtained from NCBI database (LEDGF/P75: NCBI Accession NP_001121689.1; Iduna: NCBI Accession NP_001229773.1) and codon-optimized for expression in different hosts. Sequence-based prediction online servers, PROSO (http://mbiljj45.bio.med.unimuenchen.de:8888/proso) (Smialowski et al. 2012) and GlobPlot (http://globplot.embl.de) (Linding et al. 2003), were used to optimize the solubility of chimeric E3 proteins. For eukaryotic expression, 146LI and other designed chimeras were cloned into the pEF-Flag plasmid at NheI/ MluI sites. 146LIS and its derivatives were cloned into the pEF-MYC plasmid at the NheI/MluI sites. HIV-1 NL4-3 integrase was cloned into pcDNA3.1-vector as described previously (Cherepanov et al. 2000). For E. coli expression, 146LIS was cloned into the pET30a vector at the NcoI/XhoI sites; 146LIL was cloned into the pET30a vector at the NdeI/XhoI sites; and HIV-1 NL4-3 integrase was cloned into a modified pET42a vector with the S-tag sequence deleted. For Jurkat cell knock-in, donor plasmid pBS-AAVS1-EF1α-146LIS-PURO was constructed by insertion at pBS-SK(-) XhoI/EcoRI sites of two flanking homologous arms of human AAVS1 and sequences from pEF-MYC-146LIS amplified by PCR. pX330-AAVS1-T2- CRISPR was constructed based on pX330-U6-Chimeric_BB-CBh-hSpCas9 (from Dr. Feng Zhang, Addgene # 42230) (Cong et al. 2013). The AAVS1 target sequence was described previously (Mali et al. 2013). TRIM33 (NCBI Accession NM_015906.4) sequence was amplified from a cDNA library by PCR and cloned into the pEFMYC plasmid at NheI/MluI sites.
-
293T and Jurkat cells were obtained from American Tissue Culture Collection (ATCC). Ghost-CXCR4 (X4) cells were obtained from the AIDS Reagents and Reference Program of the NIH. Primary CD4+ T lymphocytes were isolated and activated as described (Hou et al. 2015). 293T and Ghost-X4 cells were cultured in Dulbecco's modified Eagle's medium (DMEM). Jurkat cells were cultured in RPMI 1640 medium (ATCC). Culture media were supplemented with 10% foetal bovine serum (FBS). Cells were grown in a 37 ℃ humidified incubator with 5% CO2.
Jurkat 146LIS cells were generated using the CRISPR/ Cas9 system. Jurkat cells were grown in a six-well plate and two plasmids (pBS-AAVS1-EF1α-146LIS-PURO and pX330-AAVS1-T2-CRISPR) were transfected using Xfect (Takara). Two days after transfection, cells were moved to 6-cm dishes, followed by selection with 1 μg/mL puromycin. After 10–12 days, colonies were picked for further selection in a 96-well plate. The MYC-146LIS knock-in strain was verified by genome PCR and immunoblotting.
-
Genomic DNA of MYC-146LIS knock-in cells was extracted by DNA Purification Kit (Thermo). To detect integrated EF1α-146LIS-PURO sequence, a nest PCR was performed using Phanta Max Super-Fidelity DNA Polymerase (Vazyme) as manufacturer's instructions. The first round PCR used 200 ng genomic DNA as template and sequencing primers targeting EF1α and PURO sequences were used. In the second round PCR, 1 μL PCR production of the first round PCR was used as template, and primers target MYC-146LIS sequence was used.
-
Cells were washed with PBS and lysed in RIPA buffer containing a protease inhibitor cocktail (Roche). Cell lysates were sonicated before adding 2 × SDS sample buffer. After denaturation at 95 ℃ for 5 min, equal amounts of total protein were separated using SDS-PAGE. Proteins were transferred to PVDF membranes (GE Healthcare) and blotted with antibodies after blocking in 5% skim milk/TBST for 1 h at room temperature. Detection was performed using Amersham ECL Prime reagents (GE Healthcare) with a ChemiDoc Touch system (BioRad).
NP-40 buffer was used when protein extracts were prepared for the co-IP assays. Cell lysates were incubated with anti-Flag M2 Affinity Gel (Sigma) overnight at 4 ℃. The beads attached to the proteins were subsequently washed 3 times with lysis buffer, and denatured at 95 ℃ in 2 × SDS sample loading buffer for 5 min to elute the immunoprecipitates.
The antibodies used include: Flag and MYC tag antibodies (mouse) purchased from Sigma, HIV-1 integrase and ubiquitin antibodies (mouse) purchased from Santa Cruz, and α-tubulin antibody (rabbit) purchased from Sigma. MLL1, JPO2, IWS1, POGZ antibody purchased from Abcam.
-
pET30a-146LIS, pET30a-146LIL and GST-Integrase-His were expressed as soluble proteins in E. coli strain BL21(DE3) at 16 ℃. IPTG (1 mmol/L) was added at A600 ~ 0.6 and cells were harvested after 36 h. 146LIS and 146LIL proteins were purified by Ni-NTA beads (QIAGEN) according to the manufacturer's instructions. The GST-integrase-HIS proteins were purified by affinity chromatography on glutathione (GSH) beads. In general, cell pellets were frozen overnight at - 80 ℃, thawed, and resuspended in 2 mL/g of lysis buffer with protease cocktail. HIS fusion protein was absorbed onto Ni-NTA beads and washed three times with buffer containing 30 mmol/L imidazole. HIS fusion protein was eluted by buffer containing 300 mmol/L imidazole. GST fusion protein was absorbed onto GSH beads (QIAGEN) and washed three times. Factor Xa protease (50 ng/μg target protein) was used to digest the GST tag. Factor Xa was then removed by XarrestTM Agarose (Millipore).
-
The in vivo ubiquitination assay was conducted under denaturing conditions. After transfection, 40 μmol/L MG132 (Enzo Life Sciences) was added to the cell culture media 4 h prior to harvest. 36 h post transfection, cells were lysed by immediately boiling for 10 min in 1% SDS lysis buffer containing 5 mmol/L N-ethylmaleimide. The cell lysis debris was sheared with a sonication device and diluted to a final concentration of 0.1% SDS with Tris buffer. Triton X-100 was added to a final concentration of 1%. The lysates were immunoprecipitated by anti-Flag M2 Affinity Gel (Sigma) overnight at 4 ℃ and denatured at 95 ℃ in 2 × SDS sample loading buffer for 5 min to elute the immunoprecipitated proteins.
In vitro ubiquitination assays were performed by mixing 1 μmol/L of the bacterially produced recombinant proteins (146LIS, 146LIL, HIV-1 integrase) with 0.1 μmol/L E1 (UBE1, Boston Biochem), 1 μmol/L E2 (UbcH5b, Boston Biochem) and 2.5 μmol/L ubiquitin (Boston Biochem) in 25 μL of reaction buffer (50 mmol/L Tris, 5 mmol/L MgCl2, 5 mmol/L ATP, 1 mmol/L dithiothreitol, pH 7.4) according to the manufacturer's instructions. The reaction was stopped after 1 h at 37 ℃ by the addition of SDS sample buffer and boiling at 95 ℃ for 5 min.
-
The replication competent HIV-1 strain NL4-3 was produced by transient transfection of the viral vector pNL4-3 into 293T cells using PEI. The single-round infectious viral particles were produced by transient transfection of plasmid pWPI, pSPAX2 and pMD2G into 293T cells using PEI. The viral titers were measured in Ghost-X4 cells by flow cytometry (Kutner et al. 2009).
Before infection, the virus aliquots were treated with DNase I (20 U/mL, Sigma) at 37 ℃ for 1 h to remove the residual viral DNA. Jurkat and Jurkat146LIS cells were incubated with HIV-1 NL4-3 viral stock for 6 h at the indicated MOI in the presence of polybrene (8 μg/mL). Cells were washed with PBS and harvested for subsequent experiments at the indicated times.
-
PCR mixes containing 50 ng DNA, 0.2 μmol/L primers, 0.1 μmol/L probe and 1 × TaqManTM Fast Advanced Master Mix were incubated at 50 ℃ for 2 min and 95 ℃ for 15 min, followed by 40 cycles of 94 ℃ for 15 s, 58 ℃ for 30 s, and 72 ℃ for 30 s. Primers AE2963/AE4422 and probe AE2965 were used to detect late reverse transcripts. Two–LTR circles were detected using primers AE4450/ AE4451 and probe AE4452. The integration of viral DNA was measured by nested Alu-PCR. The first round of reactions used primers AE3014/AE1066, and the second round of real-time reactions used primers AE3013/AE990 and probe AE995. Standards were prepared by diluting pNL4-3.R-E-Luc or pUC18.2LTR in DNA from uninfected cells. Integration standards were made by diluting DNA from cells infected with NL4-3 virus with DNA from uninfected cells. Values obtained from parallel reactions that omitted Alu-specific AE1066 primer from first round of samples were subtracted from the Alu-PCR values obtained from the second reactions (Sowd et al. 2016).
-
The cell culture supernatants containing HIV-1 particles were collected and lysed in 0.2% Triton X-100 prepared in PBS buffer. Consecutive dilutions were made to fit the p24 concentration in the linear range of a standard curve. HIV-1 p24 ELISAs were performed using a p24 Antigen ELISA kit (Zepto Metrix) according to the manufacturer's instructions.
-
Equal amounts of WT or 146LIS knock-in Jurkat cells (5 × 103) were plated in 96-well plates and cultured for 12 h. For the cell viability assay, 10 μL CCK-8 solution (Thermo) was added to each well, and the plate was incubated at 37 ℃ for 2 h. The absorbance at 450 nm was measured using a microplate reader (Bio-Rad, PA).
-
All statistically analyzing experiments were performed in triplicate, and the data are expressed as the mean ± SD. The significance was determined by Student's t test using GraphPad Prism software. In all tests, P < 0.05 was considered statistically significant, marked as follows: *P < 0.05; **P < 0.01; ***P < 0.001.
Sequences and Plasmids
Cell Culture and Reagents
Genome PCR
Immunoblotting and Immunoprecipitation (IP)
Protein Expression and Purification
In Vivo and In Vitro Ubiquitination Assay
Virus Production and Infection
Quantitative PCR
HIV-1 p24 ELISA
CCK8 Assay
Statistical Analysis
-
In order to generate a functional chimeric E3 that can target HIV-1 integrase, the substrate-binding domains of various natural E3s have been replaced with the HIV-1 integrasebinding domain (IBD) of human LEDGF/P75 protein or the enzymatic domains of E3s were linked with the IBD directly (Fig. 1A) (Maertens et al. 2003). More than 100 chimeric E3 prototypes were generated and tested whether they can promote the degradation of HIV-1 NL4-3 integrase when co-expressed in cells. It turned out that E3 146LI (named from Iduna and LEDGF) is the only chimeric E3 which led to robust degradation of HIV-1 integrase (Fig. 1B, 1C). 146LI contained the RING domain of Iduna, which recruit E2s to process ubiquitination.

Figure 1. Construction of the chimeric E3s and functional screening. A Design of the chimeric E3s. B Diagrams of of HIV-1 integrase, host protein LEDGF/P75, and E3 Iduna domains. Colored regions denote key function domains used for chimeric E3 construction. C Representative screening data: 293T cells were co-transfected with pEFFlag-chimera and pcDNA3.1-IN, and cell lysates were immunoblotted with anti-Flag antibody, anti-integrase antibody and anti-actin antibody. Lane 1 is chimera 146LI, and lanes 2–7 are other failed candidates (not discussed in this paper) that were tested simultaneously. D Diagrams of chimeric E3s 146LI and its derivatives. E Structural modeling of chimeric E3 146LIS based structures of Iduna RING-Linker-WWE and LEDGF/P75 IBD. The Iduna RING domain is colored red. The Iduna WWE domain is colored light blue. The linker between Iduna RING and the WWE domains is colored yellow. The IBD of LEDGF/P75 is colored green.
To further optimize the activity of 146LI and to facilitate the subsequent in vitro studies, several modified versions of 146LI have been generated, including 146LIS (146LI short), 146LIS DLinker, 146LIS H37A and 146LIL (146LI long). 146LIS contained the N-terminal RING-linker domain of human E3 Iduna (Kang et al. 2011), followed by an additional flexible linker GSGSG, and IBD domain of LEDGF/P75 (Fig. 1D). The 146LIS DLinker mutant lacked the linker domain of Iduna which was proved in other failed attempts to influence chimera's activity (Fig. 1D). 146LIS H37A was an E3 activity-deficient mutant (Callow et al. 2011) (Fig. 1D). 146LIL (146LI long) has more amino acids than 146LIS in both the RING and IBD domains (Fig. 1D). All constructs were designed based on the selected domains and were codon-optimized into distinct nucleotide sequences for eukaryotic or prokaryotic expression, respectively (Fig. 1D).
Based on the structure of Iduna RING-Linker-WWE domain (PDB: 4QPL), a 3D model was simulated with its WWE domain replaced by LEDGF/P75 IBD domain (Fig. 1E). In this predicted structure of 146LIS, the size of the LEDGF/P75 IBD was similar to that of the Iduna WWE domain. The Iduna RING and linker domains formed an 'L' frame, with LEDGF/P75 IBD on the right side of the 'L' frame, revealing the spatial compatibility of 146LIS. The simulated structure suggested that the binding interfaces between RING-E2 and IBD- integrase were ipsilateral, leading to the spatial proximity of E2 and HIV-1 integrase when both bound to 146LIS. Thus, ubiquitination of HIV-1 integrase by the recruited E2 is spatially possible (Fig. 1E).
-
To screen all constructed chimeric E3s for their activity to mediate the degradation of HIV-1 NL4-3 integrase, 293T cells were transfected with the pEF-MYC-E3 and pcDNAintegrase plasmids. Intracellular integrase protein levels were examined by Western analysis. Results showed that 146LI caused obvious reduction of integrase protein level (Figs. 1C, 2A). Furthermore, co-transfection of pEF-MYC-146LIS and pcDNA-integrase into primary CD4+ T lymphocytes reinforced these results (Fig. 2B).

Figure 2. 146LIS promotes proteolytic degradation of HIV-1 NL4-3 integrase in both 293T cell lines and primary CD4+ T lymphocytes. A 293T cells were transfected with 2 μg of pEF-MYC-E3s and/or 0.4 μg of pcDNA3.1-IN with empty plasmids added to a total of 4 μg DNA. 36 h post transfection, cell lysates were immunoblotted with anti-integrase, anti-MYC antibodies, and anti-tubulin antibody. B Primary CD4+ T lymphocytes were transfected with pEF-MYC-E3s, pcDNA3.1-IN plasmids and cell lysates were immunoblotted as in (A). C Primary CD4+ T lymphocytes were transfected with pEFMYC-E3s, pcDNA3.1-IN plasmids as in (A). 40 μmol/L MG132 was added to cells 4 h prior to harvest. Cell lysates were subjected to immunoblotting with anti-integrase or anti-MYC antibodies. D Primary CD4+ T lymphocytes were transfected with 0.4 μg of pcDNA3.1-IN and increasing amounts of pEF-MYC-146LIS (0.2, 0.4, 0.8, or 1.6 μg) or pEF-MYC-146LIL (0.3, 0.5, 1.0, or 2.0 μg). Total DNA was equalized to 4 μg with the corresponding empty vectors. Cell lysates were immunoblotted as in (A).
Both 146LIS DLinker and 146LIL caused reduced integrase protein levels with weaker activity compared to 146LIS (Fig. 2A, 2B, 2D), suggesting that Iduna linker domain plays a non-critical role for chimeric E3 activity; while increased amino acids beyond RING and IBD domains are unnecessary for functional chimeric E3. As expected, the E3 activity-deficient mutant 146LIS H37A showed no effect on integrase level (Fig. 2A, 2B), indicating the enzymatic activity of 146LIS is indispensable for its activity to mediate integrase reduction. Treatment with the proteasome inhibitor MG132 could restore the protein levels of HIV-1 integrase, demonstrating that 146LIS mediated HIV-1 integrase reduction via proteasome degradation (Fig. 2C). Titration with increasing amounts of pEF-MYC-146LIS further confirmed that chimeric E3 146LIS could mediate HIV-1 integrase degradation in a dose-dependent manner (Fig. 2D). These results indicated that 146LIS and its functional variations were able to induce proteasome-dependent degradation of HIV NL4-3 integrase in human cells.
-
Our chimeric E3s were designed to interact with HIV-1 NL4-3 integrase and catalyze its Lys48-specific polyubiquitination. To confirm their interaction and subsequent substrate ubiquitination, 146LIS and its derivatives were co-transfected with pcDNA-integrase-flag into primary CD4+ T lymphocytes. After treatment with MG132, cells are harvested and co-IP experiments were done. The results indicated that 146LIS and all its derivative mutants, including the enzymatically defective mutant, could interact with HIV-1 NL4-3 integrase (Fig. 3A). Co-IP and immunoblotting performed under denaturing conditions demonstrated that 146LIS and its derivative mutants, except for the enzymatically defective mutant 146LIS H37A, could mediate the Lys48-specific polyubiquitination of integrase in vivo (Fig. 3B). These results proved that our E3 chimera 146LIS, as expected, has the ability to bind HIV-1 NL4-3 integrase and catalyze its Lys48-specific polyubiquitination in cells.
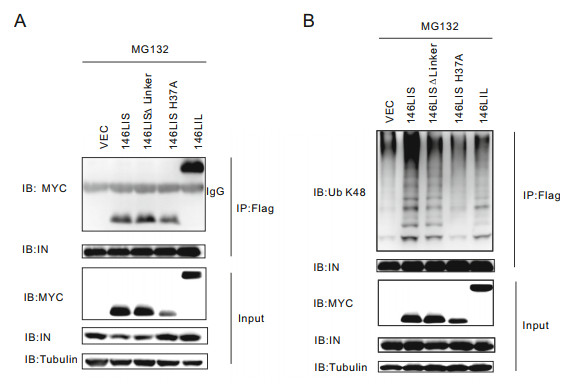
Figure 3. 146LIS interacts with HIV-1 NL4-3 integrase and mediates its Lys48-linked polyubiquitination in primary CD4+ T lymphocytes. A Primary CD4+ T lymphocytes were transfected with pEF-MYCE3s, pcDNA3.1-IN plasmids and treated with 40 μmol/L MG132 described in Fig. 2C. Co-IP experiments were carried out as described in "Materials and Methods" section. Top two panels: Immunoprecipitates were probed with anti-MYC and anti-integrase antibody. Bottom three panels: Cell lysates were probed with anti-MYC, antiintegrase antibody, and anti-tubulin antibody. B pEF-MYC-E3 (4 μg) and pcDNA3.1-IN-flag (2 μg) were co-transfected into primary CD4+ T lymphocytes. The in vivo ubiquitination of integrase was analyzed by Flag IP under denaturing conditions as described in "Materials and Methods" section. Top two panels: Immunoprecipitates were probed with Lys48-specific ubiquitin chain antibody and anti-integrase antibody. Bottom three panels: Cell lysates were probed with antiMYC, anti-integrase antibody, and anti-tubulin antibody.
-
Considering overexpressed HIV-1 integrase may have background ubiquitination in eukaryotic cells (Mousnier et al. 2007; Ali et al. 2019) (Fig. 3B), in vitro ubiquitination assay was employed to further confirm the E3 activity of our chimeric E3s. In order to avoid contaminations from other mammalian E3s, Escherichia coli (E. coli) expression system was used to express the chimeric E3s and substrate integrase. However, our initial successful screening product, E3 chimera 146LI, was failed to be expressed as soluble form in E. coli. Therefore, 146LI derivatives have been designed and further analyzed. Finally, HIV-1 integrase and functional E3 chimera alternates 146LIS and 146LIL have been successfully expressed in E. coli and purified.
To examine their abilities to ubiquitinate substrate integrase in vitro, prokaryotically expressed 146LIS or 146LIL was assayed with or without E1, E2 UbcH5b, ubiquitin and integrase-His at 37 ℃ for 60 min in the presence of ATP. E2 UbcH5b was chosen because it has previously been used as the cognate E2 for Iduna in vitro (Kang et al. 2011). Chimeric E3 146LIS showed autoubiquitination activity in the presence of E1, E2 and ubiquitin (Fig. 4A, lane 2), and displayed strong E3 activity as it promoted robust integrase polyubiquitination (Fig. 4A, 4C, lane 5). Without E3 chimeras, E1, E2 and ubiquitin themselves could not activate integrase polyubiquitination (Fig. 4A–4C, lane 1). Furthermore, 146LIS specifically promoted the Lys48-linked polyubiquitination of integrase, as shown by immunoblotting using an antibody specific for Lys48-linked polyubiquitin chains (Fig. 4C). Since the ability to autoubiquitinate themselves and to ubiquitinate substrate proteins are characteristics of ubiquitin E3 ligases (Fuchs et al. 2002), our in vitro data further confirmed that the chimeric E3 146LIS is a fully functional E3 ligase which can specifically mediate the Lys48-linked polyubiquitination of HIV-1 integrase in vitro.

Figure 4. In vitro ubiquitination of HIV integrase promoted by 146LIS (A, C) or 146LIL (B). 146LIS, 146LIL, and integrase proteins were purified and employed in in vitro ubiquitination experiments as described in "Materials and Methods" section. 146LIS or 146LIL was assayed in the presence or absence of E1/E2, integrase and ubiquitin (Ub) as indicated. Samples were analyzed by immunoblotting against Ub (A, B) or Lys48-specific ubiquitin chains (C).
Lys48-linked polyubiquitination is considered to be the key posttranslational modification for protein degradation via the UPP (Ciechanover 1994). Our in vitro results implied that 146LIS would have the ability to mediate substrate degradation of HIV-1 integrase in vivo, consistent with our previous in vivo studies. And also being consistent with the appeared weaker activity in mediating integrase polyubiquitination and degradation in vivo (Figs. 2A, 2B, 2D, 3B), the longer version 146LIL showed even more obviously reduced integrase polyubiquitinating activity in vitro compared to 146LIS (Fig. 4B). 146LIL-mediated polyubiquitinated integrase even is too little to be detected by the antibody against Lys48-specific polyubiquitin chains (data not shown). Therefore, it is concluded that sequence changes beyond the vital functional domains could also influence the chimeric E3 activity. The ability of chimeric E3s to mediate integrase polyubiquitination and degradation in cells is directly related to their substrate polyubiquitinating activity in vitro.
Similar to 146LI, 146LIS ΔLinker could not be expressed as soluble protein in E. coli, implying that the linker domain of Iduna may play roles in stabilizing protein conformation and facilitating enzymatic activity of E3.
-
As a functional E3 ligase specifically promotes degradation of HIV-1 NL4-3 integrase, 146LIS was presumed to function as an intracellular inhibitor of HIV-1 by interfering HIV-1 genome integration. To prove this hypothesis, Jurkat cells with 146LIS knock-in (Jurkat146LIS) were generated by an approach based on sequence replacement by CRISPR/Cas-mediated homology-directed repair (HDR) (Liang et al. 2015; Merling et al. 2015). 146LIS donor plasmid pBS-EF1α-146LIS-PURO was constructed using pBS-SK(-) vector inserted by consecutive sequences of flanking AAVS1 homology arms (750-bp sequences of adeno-associated virus integration site 1 from human genome), EF-1a promoter, codon-optimized 146LIS gene, internal ribosome entry site (IRES), puro resistance gene and SV40 poly(A) terminator (Hobbs et al. 1998; Natsume et al. 2016) (Fig. 5A). After co-transfection of pBS-EF1α-146LIS-PURO and pX330-AAVS1-T2-CRISPR into Jurkat cells and puromycin selection, 146LIS gene insertion was verified by genomic PCR (Fig. 5B) and MYC-146LIS protein expression was confirmed by Western blotting (Fig. 5C).

Figure 5. 146LIS knock-in Jurkat cells restrain HIV-1 viral replication. A Experimental scheme used to introduce EF1α-146LIS at the AAVS1 locus of human cells. Cells were transfected with pBS-EF1α- 146LIS-PURO and pX330-AAVS1-T2-CRISPR to target the AAVS1 locus, followed by puromycin selection as described in "Materials and Methods" section. B After selection, 4 clones were tested for the integrated MYC-146LIS gene (0.5 kb) by genomic nested PCR. C Immunoblotting to detect 146LIS protein expression in Jurkat146LIS knock-in cells. The indicated clones were assayed by immunoblotting using an anti-MYC antibody. D Jurkat and Jurkat146LIS cells were infected with HIV NL4-3 at MOIs of 0.1 and 1. Five days after infection, cell supernatants were harvested to measure viral replication by p24 ELISA. **P < 0.01; ***P < 0.001. E Jurkat and Jurkat146LIS cells were challenged with single-round infectious viral particles at MOI of 5. Six hours after infection, cell lysates were immunoblotted as in Fig. 2A. F Jurkat and Jurkat146LIS cells were transfect with pNL4-3 plasmid. 48 h after infection, cell lysates were immunoblotted as in Fig. 2A.
In order to investigate the antiviral efficiency of 146LIS knock-in, Jurkat and Jurkat146LIS cells were exposed to HIV-1 NL4-3 virus at an MOI of 0.1 or 1, respectively. Five days post virus infection, the p24 levels in Jurkat146LIS were approximately ten-fold lower than those in WT Jurkat cells at an MOI of 0.1 and five-fold lower at an MOI of 1 (Fig. 5D), demonstrating that 146LIS knock-in could significantly restrained viral replication in Jurkat cells.
To figure out whether integrase was degraded upon entry of the virus or during assembly, we monitored the protein levels of integrase in Jurkat146LIS cells during virus entry and viral production. Jurkat and Jurkat146LIS cells were challenged with single-round infectious viral particles at an MOI of 5. Six hours after infection, it showed that integrase protein level was significant decreased in Jurkat146LIS cells, indicating that integrase was degraded upon entry of viral particles (Fig. 5E).Similarly, 48 h after pNL4-3 plasmid transfection of Jurkat and Jurkat146LIS cells, the cellular integrase protein level in Jurkat146LIS cells was also significant decreased, indicating that integrase was degraded during new viral particles production (Fig. 5F). Therefore, the viral integrase is degraded upon both entry of the virus and viral assembly.
To further analyze whether the antiviral effect brought by 146LIS knock-in is due to inhibition of HIV-1 DNA integration, cellular DNA isolated from the infected cells was quantitatively analyzed for viral DNA metabolism by real-time fluorescence PCR (Fig. 6). The results showed that late RT products reached maximum level at 12 h after infection while no significant difference observed between Jurkat146LIS cells and Jurkat WT cells (Fig. 6A, 6B). Less 2-LTR products were detected in Jurkat146LIS cells at an MOI of 0.1, but no significant difference was observed at an MOI of 1 (Fig. 6C, 6D). These data indicated that 146LIS had little effect, as we expected, on unintegrated viral DNA synthesis (linear and circular). Impressively, 24 h after infection more than ten-fold less integrated viral DNA was detected in Jurkat146LIS cells at an MOI of 0.1 and approximately five-fold less integrated viral DNA was detected at an MOI of 1(Fig. 6E, 6F), showing that 146LIS knock-in lymphocyte cells obtained a strong ability to resist viral DNA integration during HIV-1 NL4-3 infection.

Figure 6. 146LIS knock-in shows little effect on unintegrated viral DNA but strong inhibition on integrated viral DNA. Jurkat WT and 146LIS were exposed to HIV-1 NL4-3 at MOIs of 0.1 and 1. Cells were harvested at the indicated time points for DNA extraction and quantified by subsequent real-time PCR for late RT levels (A, B), 2-LTR-circle levels (C, D), and integrated DNA levels (E, F). **P < 0.01; ***P < 0.001.
-
To examine whether the expression of chimeric E3 146LIS would affect cell growth, cell viability assay was performed using Cell Counting Kit-8 (CCK8). Results showed that Jurkat146LIS cells grew similarly to Jurkat WT cells (Fig. 7A), indicating the expression of chimeric E3 146LIS applied no effect on cell growth.

Figure 7. Cytotoxicity and cellular protein disturbance tests for 146LIS. A Cell viability of Jurkat WT and 146LIS knock-in was determined by the CCK8 assay as described in "Materials and Methods" section. B pcDNA3.1-IN plasmid was co-transfected with pEF-MYC-146LIS or pEF-MYC-TRIM33 plasmids into Jurkat cells as in Fig. 2A. Cell lysates were immunoblotted with anti-integrase antibody, anti-MYC antibody and anti-tubulin antibody. C Jurkat cells were transfected with 4 μg pEF-MYC-146LIS plasmid. Cell lysates were immunoblotted with anti-MLL1 antibody, anti-JPO2 antibody, anti-IWS1 antibody or anti-POGZ antibody. D Morphology of Jurkat WT and 146LIS knock-in after culturing for 6 months under the same condition. Scale bar = 100 μm.
A few natural E3 ligases were also shown to target HIV integrase for degradation (Ali et al. 2019). Thus, TRIM33 was cloned as a representative to be compared with 146LIS in terms of efficiency. Jurkat cells were transfected with pcDNA3.1-IN plasmid and the pEF-MYC-E3 plasmids. Intracellular integrase protein levels showed that the degradation efficiency of 146LIS was much higher than natural E3 ligase TRIM33, consistent with the fact that 146LIS is an engineered E3 ligase specifically designed for efficient integrase degradation while TRIM33 is a host natural ubiquitin ligase mainly targeting host proteins (Fig. 7B).
Although 146LIS only contains the integrase-binding domain of LEDGF/P75 protein, LEDGF/P75 could also interact with other cellular proteins such as IWS1 (Tesina et al. 2015), POGZ (Bartholomeeusen et al. 2009), MLL1 (Yokoyama and Cleary 2008) and JPO2 (Maertens et al. 2006). To assure 146LIS won't target these proteins for degradation, 146LIS was transiently expressed in Jurkat cells and these protein expressing levels were examined by immunoblotting. Results showed that 146LIS affected the protein levels of none of these proteins in cells (Fig. 7C), suggesting 146LIS might have high specificity for HIV-1 integrase.
Cell growth and morphologies of Jurkat146LIS and Jurkat WT cells were continuously observed through the whole culture period of 6 months with no obvious change observed (Fig. 7D). Taken together, we concluded that 146LIS expression did not affect lymphocyte cell growth and has no obvious cell cytotoxicity.
Construction of Chimeric E3s, Functional Screening and Generation of E3 146LIS and Other Derivatives
146LIS Promotes Proteolytic Degradation of HIV-1 NL4-3 Integrase in Both 293T Cell Lines and Primary CD4+ T Lymphocytes
146LIS Interacts with HIV-1 NL4-3 Integrase and Mediates Its Lys48-Linked Polyubiquitination in Primary CD4+ T Lymphocytes
146LIS Specifically Promotes Lys48-Linked Polyubiquitination of HIV Integrase In Vitro
146LIS Knock-in Jurkat Cells Restrains HIV-1 DNA Integration and Viral Replication
Cytotoxicity and Cellular Protein Disturbance Tests for 146LIS
-
The chimeric E3 146LIS is a promising attempt for developing new anti-HIV strategies. 146LIS was designed in a continuous translational term in order to utilize CRISPR knock-in technique, therefore sidestepped the potential transmembrane problems of many molecular tinkering drugs. In our follow-up research, the abilities of 146LIS to resist DNA integration of other HIV-1 strains, and the effects of 146LIS on other integrase interacting proteins would also be investigated. An ultimate optimized 146LIS gene expression sequence could be integrated into genome of isolated hematopoietic stem cells at AAVS1 safe harbor site in vitro. These modified hematopoietic stem cells could then be transplanted back to the donor patients, producing 146LIS transgene differentiated cells. This approach may produce sufficient quantity of HIV-1 DNA integration resisting cells, thereby realizing hopeful gene therapy of AIDS with the combined use of other HIV- 1 suppression drugs.
The IBD and RING domain of 146LIS were obtained from human proteins, which makes 146LIS unlikely to induce host immune responses. Our initial cytotoxicity experiments showed that 146LIS affected neither cell growth nor the morphology of Jurkat cells. But the toxicity is still the primary concern before this gene therapy approach could be safely applied to any clinical practice. Questions remain that whether the permanent expression of an engineered ligase throughout haematopoietic development affect any host protein and be harmful for any of the haematopoietic stem cell progeny. In our subsequent study, we will generate 146LIS knock-in hematopoietic stem cells and perform a comprehensive investigation of potential toxicity. Further analysis like RNA seq analysis and proteome analysis of cells with or without HIV infection will be employed. In addition, the current version of our chimeric ubiquitin ligase is a result of multiple iterations, and more attempts are worthwhile to try to maximize degradation efficiency and minimize the side effects to human cells.
Integration of viral genetic information into the host genome is a unique step during lifecycle of the retrovirus (Engelman et al. 2009), which is carried out by integrase (IN). IN catalyzes two important enzymatic reactions, 3' processing of provirus DNA (Hare et al. 2012) and cleavage of host chromosomal DNA for transfer of the viral DNA (Bushman and Craigie 1991). Binding of HIV-1 integrase to host chromatin relies on the interaction between integrase and the host chromosome anchoring protein LEDGF/P75. Targeting on integrase/integration, two antiviral strategies have been developed. Integrase strand transfer inhibitors (INSTIs) bind the divalent metal ions and viral DNA end after 3' processing, preventing the subsequent strand transfer reaction (Pommier et al. 2005). Allosteric integrase inhibitors (ALLINIs) inhibit the interaction of integrase with LEDGF/P75, thus reducing HIV-1 integrase activities in a LEDGF/P75-dependent manner (Christ et al. 2010). However, the mutation rate of retroviruses is very fast. Even a single amino acid mutation is enough to cause HIV-1 integrase resistance to antiviral drugs (Cooper et al. 2008; Sharma et al. 2014). Consequently, antiviral research on HIV-1 is an ongoing and challenging task. Our artificial chimeric E3 146LIS exploits the modular architecture of a conservative hostvirus protein interaction to develop a new anti-viral approach. The critical role of the host protein LEDGF/P75 IBD in the HIV-1 lifecycle has been previously addressed. When clinical HIV-1 strains were used, sterilizing infections were observed in LEDGF/P75 KO cells (Schrijvers et al. 2012). This interaction is too critical for virus to be targeted by virus mutations. If virus mutations cause integrase unable to bind LEDGF/P75 IBD, viral DNA integration will also fail. Thus, the design of 146LIS took advantage of this critical interaction between integrase and LEDGF/P75 IBD, making 146LIS a reasonable approach to avoid the disadvantages of virus mutations.
Some previously successful approaches to build a new E3 for targeting a novel substrate are not suitable for the purpose of this study due to several problems, including introducing macromolecular ligands synthesized in vitro into the cell, challenges for passing through cell membranes, and usage of foreign substances which may activate the host immune response. To achieve our goal to develop feasible chimeric E3s which may safely protect people from HIV infection, the following criteria need to be satisfied: (1) To ensure chimeric E3 can be used in gene therapy, it should be coded by a continuous DNA sequence. (2) The selected E3s should be capable of mediating Lys48-specific ubiquitination of its target both in vivo and in vitro. (3) The efficiency of enzymatic degradation should be sufficiently high to avoid escape of viral enzymes. (4) The resulting transcript and protein should be harmless to host cells. Many similar studies have claimed a universal method to degrade diversified protein substrates. However, in our study, successful Lys48-specific ubiquitination of the integrase by an engineered E3 requires the right combination of the substrate binding region and E3 enzymatic domain. After choosing LEDGF/P75 IBD as the HIV-1 integrase-binding region, we tested enzymatic domains of many different E3s. However, few enzymatic domains were able to target HIV-1 integrase when linked with the LEDGF/P75 IBD. After a large-scale screening, we found that the Iduna RING domain met the criteria when placed in the N-distal region of 146LIS. Unlike most other E3s, the RING domain of Iduna is located in the N-end of this protein. Thus, changing the relative positions of the RING domain and IBD might be important for the E3 activity achieved by chimeras. In our attempts, none of the tested HECT family E3s, or other RING E3s, U-BOX E3s, Cullin-based E3s, could produce a functional chimeric E3 to degrade HIV-1 integrase, suggesting that the construction of a functional E3 chimera is not simple.
In this study, we proved that it is feasible to construct chimeric ubiquitin ligases to target a viral protein. The design of chimeric E3 146LIS could be borrowed to generate more functional E3 chimeras to target alternate HIV viral proteins as well, or applied to new strategies which can protect people from other severe viruses and human diseases.
-
This work is supported by Grants from the National Science and Technology Major Project of China (2014ZX-10001003), National Natural Science Foundation of China (#81620108020 & #31400774).
-
MW conceived the strategy of this study. ZZ designed the chimeric E3s and performed the screening, experiments and the analyses. SY and SX took part in experiments. DG, LC and WH gave advice for experiments. ZZ and MW wrote the manuscript. DG revised the manuscript. All authors have read and approved the contents of the final manuscript.
-
The authors declare that they have no conflict of interest.
-
This article does not contain any studies with human or animal subjects performed by any of the authors.


 DownLoad:
DownLoad: