











-
Epstein-Barr virus (EBV) is a human gamma herpes virus closely associated with nasopharyngeal carcinoma (NPC). Among the twelve proteins encoded by the EBV genome, the latent membrane protein-1(LMP1) gene has been a focus of interest as a target gene due to its ability of transform certain rodent fibroblast cell lines (3). Therefore, triggering RNAi (RNA interference), by the introduction of siRNA (small interfering RNA) aimed at the LMP1 gene, into NPC cells to knockdown LMP1 gene expression holds promise as a gene therapy approach for control of NPC.
siRNA is most commonly achieved in stable form by using shRNA (short hairpin RNA) expressed from a DNA vector or virus (5, 6, 8, 10) with a polymerase-Ⅲ U6/H1 RNA promoter. We designed the LMP1 gene-specific insert (pshLMP1 expression cassette) such that it specifies a 19nt sequence derived from the target LMP1 transcript, separated by a loop of four oligonucleotides from the reverse complement of the same 19nt sequence (1). The pshLMP1 expression cassette was incorporated according to the traditional "annealing method" (1) and then cloned into the pTZU6+1 vector between the U6 promoter and T5 termination and checked by sequencing. In our previous research, deletion and mutant of nucleotides in pshLMP1 expression cassette were frequently observed when the "annealing method" was implemented, which may severely restrict its application in RNAi technology. We therefore established an alternative and novel "PCR method" to construct pshLMP1 vectors and demonstrated it has greater efficiency compared to the annealing method, and can facilitate the effective application of shRNA vectors for antiviral purposes.
HTML
-
Plasmid pTZU6+1 (a gift of Key Laboratory of Molecular Biology on Infectious Diseases of Chongqing University of Medical Sciences) containing a U6 promoter and Sal Ⅰ/Xho Ⅰ Xba Ⅰ cloning sites, was used as backbone plasmid to construct pshLMP1 vectors using both the annealing and PCR methods. The U6 promoter has a well defined start of transcription and a termination signal consisting of five thymidines in a row (T5). Plasmid pEGFP-N1-1158, an expression vector encoding LMP1 and EGFP fusion protein, was constructed in our previous work and had been functionality demonstrated by its expression of green fluorescence in cells.
HNE1 cells, a nasopharyngeal carcinoma cell lines with a negative LMP-1 gene, were chosen as the target cells to be cultured in RPMI-1640 (GibcoBRL Co, USA) supplemented with 10% heat-inactivated fetal bovine serum and 1% antibiotics solution(GibcoBRL Co, USA).
-
EBV LMP1 gene consists of three exons: a, b, c. An series of internal repeat(IRs) with a length of 35bp exists in exon c from base 747 to 920 as follows: 5'GTCCTGGGACTGTTGTGACTACTGTTACCGGGTGT3'. According to the design strategy of siRNA, the bases from 3 to 21 in this 35bp IRs were selected as the 19nt coding sequence specially targeting the LMP1 gene and were analyzed by BLAST to ensure that they did not have significant sequence homology with other genes.
-
The pshLMP1 vectors for specific LMP1-targeted shRNA have a pTZU6+1 backbone with an inserted pshLMP1 expression cassette between Sal Ⅰ/Xho Ⅰ and Xba Ⅰ cloning sites. The pshLMP1 expression cassette was designed as follows: the 19nt coding sequence from the target LMP1 transcription was separated by 4 oligonucleotides from the reverse complement of the same 19nt sequence, and by five thymidines (T5) with half or complete Sal Ⅰ/Xho Ⅰ and Xba Ⅰ sites on the 5' ends respectively. It was then obtained by one of the two following methods.
In the first "annealing method" also known as the conservative method, the forward and reverse strands were chemically synthesized and then annealed and inserted into pTZU6+1 vector (pshLMP1).
A second design involving the "PCR method" used two primers complementary both to the 5'end and 3' end of the pshLMP1 expression cassette with complete Sal Ⅰ and Xba Ⅰ sites (Fig. 1). The two primers must be matched at the 3' ends with a length of 10bps to enhance the annealing probability at the very first PCR cycle. The two primers were chemically synthesized and then used as primers and templates simultaneously in the PCR cycles. The PCR was performed as follows: 10×Taq buffer 2.5μL, MgCl2 (25mmol/L) 2 μL, dNTPs (2.5 mmol/L each) 2 μL, forward and reverse primers (20uM) 9 μL respectively, Taq polymerases 5U, up to 25 μL. The first cycle was incubated at 94℃ for 1min, 94℃ for 20s, 45℃ for 20s, 72℃ for 20s; then 30 cycles of incubation at 94℃ for 20s, 60℃ for 20s, 72℃ for 20s, and extended at 72℃ for 10min, then incubated at 4℃. After precipitation by ethanol and digestion with Sal Ⅰ and Xba Ⅰ, the amplified DNA double strands were cloned into the Sal Ⅰ-Xba Ⅰ site of pTZU6+1 vector (pshLMP1).
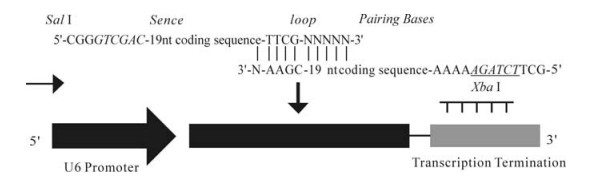
Figure 1. Strategy of PCR method. Two primers complementary both to the 5'end and 3' end of the pshLMP1 expression cassette with complete Sal Ⅰ and Xba Ⅰ sites. The vertical lines indicate the 10 bases match between two primers at 3' ends.
-
The pshLMP1 vectors obtained from both methods were transformed into competent bacteria. PCR were performed on the isolated bacteria fluid with general primers T7P (TAATACGACTCACTATAGGG) and M13F (TGTAAAACGACGGCCAGT) and pTZU6+1 vectors were taken as controls. DNA sequencing was done at Key Laboratory of Molecular Biology on Infectious Diseases of Chongqing University of Medical Sciences.
-
pshLMP1 expression vectors obtained by the PCR method were co-transfected with pEGFP-N1-1158 into HNE1 cells. The HNE1 cells were cultured in a 24-well culture vessel to a cell density of 80-90%. Then pEGFP-N1-1158 and pshLMP1 vectors were cotransfected into HNE1 cells by lipofectamine2000 (a lipid product of invitrogen) with a quality ratio of 1: 1, 1: 2 and 1: 5.
-
24 h after transfection, green fluorescence protein (GFP) was analyzed through fluorescence microscope as indicated by the expression level of LMP-1 protein in HNE1 cells.
-
24 hours after transfection, the HNE1 cells were collected and processed to obtain total mRNA and proteins. Total mRNA of HNE1 cells was extracted using standard Trizol RNA isolation protocol, followed with RT-PCR (a TOYOBO kit)to detect the mRNA levels of the LMP-1 gene (primers of LMP-1 gene were 5'-GGAACCAGAAGAGCCCCAAA-3' and 5'-CCCTCAACAAGCTACCGAT-3', with a product length of 500 bp). The proteins of HNE1 cells were extracted using M-PER mammalian protein extraction reagent, a product of PIERCE Biotechnology, and then resolved on 12% SDS-polyacrylamide gels, transferred onto nitrocellulose membranes, and incubated with the appropriate antibodies. The anti-LMP1 antibody was obtained from Dako Cytomation and used at a 1/100 dilution.
Plasmids and cell
Selection of target gene fragment
Design and construction of plasmid
Detection of recombination pshLMP1 vectors
Co-transfected with pshLMP1 and pEGFP-N1-1158 into HNE1 cellls
Green fluorescence analysis
Detection of mRNA and protein levels of LMP-1 gene
-
To confirm that the pshLMP1 expression cassettes were obtained in two methods, the double stranded hybrids purified from both "annealing method" and "PCR method" were detected by 2.5% agarose gel electrophoresis with their respective single strand DNA used as controls. Specific double-stranded DNA was obtained by both two methods (Fig. 2).

Figure 2. Detection of both double-stranded and single-stranded nucleotides by annealing and PCR methods. The double-stranded DNA indicates the presence of the expression cassette. Lane 1 (single) and lane 2 (double) show strands detected by PCR method; Lane 4 (double) and the lane 5 (single) show strands detected by annealing method.
-
After insertion of the pshLMP1 expression cassette into pTZU6+1 vectors, the PCR of recombination pshLMP1 vectors from individual colonies were performed by primers T7P and M13F to determine whether the cassettes were cloned into the vectors correctly. DNA fragments with a predicted length of 480bp were obtained by PCR (Fig. 3). Comparison with the controls of pTZU6+1 vectors, indicated the pshLMP1 expression cassette was cloned into pTZU6+ 1 for both two methods.

Figure 3. Detection of recombination pshLMP1 vectors by PCR. Lane 1 and 6, pTZU6+1 vectors, lane 2 and 5, DNA marker, 3, pshLMP1 by annealing method, 4, pshLMP1 by PCR method. The DNA fragments amplified from bacteria fluid indicate that pshLMP1 expression cassettes were correctly inserted into pTZU6+1 vectors in both two methods.
-
Verifying the sequence of pshLMP1 is essential since mismatch, deletion or mutant of even only one nucleotide within the target sequence can inhibit knockdown of the LMP1 gene (4, 9). Sequencing results for both of the two recombination pshLMP1 vectors constructed by "annealing method" and "PCR method" are shown in Figure 4. DNA sequencing peaks and alignment showed that deletion and mutant of nucleotides frequently occurred in the "annealing method" with incorporation of errors in all three sequenced (two deletions and one mutations). On the other hand the sequenced "PCR method" samples didn't contain a single deletion or mutation (Fig. 4).

Figure 4. Sequencing and alignment result of recombination pshLMP1 vectors. The arrows in bold indicate deletion and mutant of nucleotides in the pshLMP1 expression cassette. This phenomena occurs frequently with incorporation of mistakes in all three samples obtained via the annealing method. Conversely, PCR method has a better cloning efficiency as no mismatched nucleotides was found in any of the sequenced samples.
-
Compared with the positive group, GFP levels were significantly down-regulated in each treatment group (P < 0.05) and the group of pEGFP-N1-1158 (0.5μg: pshLMP1 1.0μg) had the most efficient downregulating capacity among all the treatment groups (P < 0.05) (Fig. 5).

Figure 5. The green fluorescence protein analysis for 4 test groups. A: Positive control. B: 0.5μg pEGFP-N1-1158 +0.5μg pshLMP1. C: 0.5μg pEGFP-N1-1158 + 1.0μg pshLMP1. D: 0.5μg pEGFP-N1-1158 + 2.5μg pshLMP1. Among the three treatment groups the pEGFP-N1-1158 0.5μg: pshLMP1 1.0μg group has the most efficient inhibition capacity against the LMP-1 gene in HNE1 cells (P < 0.05).
-
The mRNA levels of LMP-1 gene in the 4 groups were detected followed protocol given in the manual of the RT-PCR kit (TOYOBO inc). Compared to the positive control, the mRNA levels of the LMP-1 gene in all treatment groups were down-regulated and the group of pEGFP-N1-1158 0.5μg: pshLMP1 1.0μg had the most efficient inhibition capacity of LMP-1 gene which was consistent with the results from green fluorescence analysis (Fig. 6). The protein levels of LMP-1 gene in all treatment groups were degraded and the group of pEGFP-N1-1158 0.5μg: pshLMP1 1.0μg had the most efficient inhibition capacity (Fig. 7).

Figure 6. Detection of LMP-1 mRNA. Lane 1, DNA marker; 2, 0.5μg N1-1158+1.0μg pshLMP1; 3, 0.5μg N1-1158+2.5μg pshLMP1; 4, 0.5μg N1-1158+0.5μg pshLMP1; 5, The target DNA fragment of LMP-1 gene (500bp) and the G3PDH gene fragment (450bp) was used as control.

Figure 7. Detection of LMP-1 protein. Lane 1, Protein marker; 2, 0.5μg N1-1158 + 1.0 μg pshLMP1; 3, 0.5μg N1-1158 + 2.5 μg pshLMP1; 4, 0.5μg N1-1158+ 0.5 μg pshLMP1; 5, Positive control. The MW of LMP-1 is 55kDa and β-actin is 43kDa.
Detection of the pshLMP1 expression cassette
PCR
Sequencing of pshLMP1 vectors
Green fluorescence analysis
Detection of mRNA and protein of LMP-1 gene
-
In recent years, RNA interference has emerged as a major regulatory mechanism in eukaryotic gene expression (2). Moreover, RNAi has been evolved into a powerful tool for artificially modulating gene expression through the introduction of shRNAs. The latent membrane protein-1 encoded by Epstein Barr virus is currently of great interest for application in gene th erapy for nasopharyngeal carcinoma because of its significant role as an oncogene in NPC. In our previous studies, we were focusing on applying pshLMP1 vectors into NPC cell lines which express LMP1 and observing its inhibition efficiency against the LMP1 gene.
The traditional "annealing method" was used to construct the pshLMP1 expression cassette as described above. Unexpectedly, deletion and mutation of nucleotides were frequently found in sequencing and alignment analysis. In the traditional annealing method, the double-strands oligomers of pshLMP1 expression cassette is 60-70bp long which increases the likelihood of incorporation of higher ratios of deletions and mutations of nucleotides during the chemical synthesizing procedure. Since siRNA can have "off-target" effects, it is important for correct coding sequence to ensure the introduction of the proper siRNAs. Therefore, the errors incorporated by annealing method may severely impair the applications of RNAi technology in gene therapy of NPC.
In order to facilitate the application of shRNA technology in LMP1gene targetting, we optimized the construction process of pshLMP1 expression cassette with a novel "PCR method". The chemically synthesized oligonucleotides taken as both primers and templates in this method are of 35bp long, which is easier to obtain with a lower ratio of mismatch, deletion or mutant compared to the old annealing method. However, it is worth noting that the condition of PCR cycles is different from the traditional protocols since the method is designed to guarantee the high yield of double-stranded DNA oligomers. In the very first cycle, 45℃ was used as annealing temperature to increase the output of double-stranded DNA; in the following cycles, 60℃ was chosen to enhance the specificity of double-stranded DNA.
In our functional studies, we demonstrated that pshLMP1 expression vectors have the capacity of LMP-1 gene in HNE1 cells and discovered that pEGFP-N1-1158 0.5μg: pshLMP1 1.0μg group is of most efficient inhibition capacity which may provide a novel genetic therapy method in clinical treatment of nasopharyngeal carcinoma. In one study concerning RNA interference in hepatitis B virus expression, a combination of 4μg HBV expression vector and 15μg shRNA expression vector showed the most effective inhibition efficiency of HBV(7). In our previous research, fatality of HNE1 cells increased rapidly when pEGFP-N1-1158: pshLMP1 was present in the ratio of 1: 2 and maximum green fluorescence was gained when pEGFP-N1-1158 was about 0.5μg. Lipofectamine 2000 (Invitrogen, Calsbad CA, USA) was used as the transfection reagent which had aggravated cytotoxicity as doses of lipofectamine2000 increased. Accordingly, a combination of pEGFP-N1-1158 0.5μg: pshLMP1 1.0μg was chosen as the co-transfection doses in the functional studies in HNE1 cells. In our research, the "PCR method" provides an easier and more convenient way to construct special shRNA vectors with a higher success ratio and which are efficient in down-regulating expression of the target gene of EBV-LMP1. This novel method should be of significant use in the design and preparation of functional shRNAs tar-geting specific virus and genes.

 DownLoad:
DownLoad: